
เนื้อหา
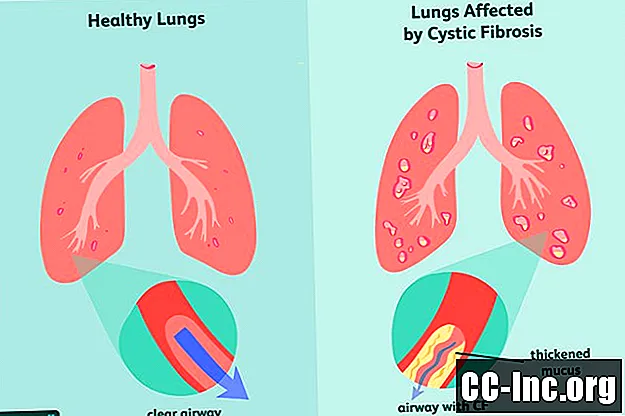
Cystic fibrosis (CF) เป็นโรคที่สืบทอดและคุกคามถึงชีวิตซึ่งทำลายปอดและทางเดินอาหาร เกิดจากยีนบกพร่องที่กระตุ้นการผลิตเมือกข้นซึ่งอุดตันทางเดินหายใจและขัดขวางการหลั่งเอนไซม์ย่อยอาหารอาการจะเกิดขึ้นเรื่อย ๆ และมักจะรุนแรงและอาจรวมถึงปัญหาการหายใจการติดเชื้อในปอดที่เกิดขึ้นอีกการเจริญเติบโตไม่ดีภาวะมีบุตรยากในเพศชายและการอักเสบเรื้อรังของตับอ่อนตับไตและหัวใจ
CF สามารถวินิจฉัยได้ด้วยการตรวจเลือดการคัดกรองทางพันธุกรรมและขั้นตอนที่เรียกว่าการทดสอบคลอไรด์ในเหงื่อ
แม้ว่าจะไม่มีการรักษา CF แต่ก็มีวิธีการรักษาที่สามารถปรับปรุงทั้งความยาวและคุณภาพชีวิตของตนเองได้
ซึ่งรวมถึงเทคนิคการล้างทางเดินหายใจยาปฏิชีวนะที่สูดดมทินเนอร์เมือกเอนไซม์ตับอ่อนอาหารที่มีแคลอรีสูงและยารุ่นใหม่ที่เรียกว่าตัวปรับ CFTR ในกรณีที่รุนแรงอาจจำเป็นต้องปลูกถ่ายปอด
อาการ Cystic Fibrosis
ในฐานะที่เป็นความผิดปกติทางพันธุกรรม cystic fibrosis เป็นสิ่งที่คุณเกิดมา อาจมีหรือไม่มีอาการในช่วงแรกเกิดและมักใช้เวลาหลายเดือนหรือหลายปีก่อนที่อาการเจ็บป่วยจะปรากฏขึ้น เมื่อถึงเวลานั้นปอดและทางเดินอาหารอาจได้รับความเสียหายที่ไม่สามารถแก้ไขได้
สัญญาณและอาการเริ่มแรกที่พบบ่อยที่สุดของ CF ได้แก่ :
- การอุดตันของอุจจาระแรกของทารก (ขี้เทา)
- ผิวรสเค็ม
- ไอเรื้อรังหายใจไม่ออกหรือมีเสมหะสี
- อุจจาระหลวมเยิ้มและมักมีกลิ่นเหม็น
- การติดเชื้อในปอดมักเกิดขึ้นอีก
- การเติบโตที่ไม่ดีและความล้มเหลวในการเจริญเติบโต
หากไม่สามารถควบคุมอาการเหล่านี้ได้ความเครียดในปอด (และไม่สามารถรับน้ำหนักได้) อาจมีผลสะสมส่งผลต่ออวัยวะหลายส่วนและเพิ่มความเสี่ยงของภาวะแทรกซ้อนของโรค
ภาวะแทรกซ้อนที่มีลักษณะเฉพาะเพิ่มเติม ได้แก่ :
- วัยแรกรุ่นล่าช้า
- Bronchiectasis (ความหนาของผนังปอดเรื้อรัง)
- ลดน้ำหนัก
- ตับอ่อนอักเสบ (การอักเสบของตับอ่อน)
- ชายมีบุตรยาก
- ความดันโลหิตสูงในปอด (ความดันโลหิตสูงในปอด)
- โรคนิ่ว
- โรคเบาหวานที่เกี่ยวข้องกับโรคซิสติกไฟโบรซิส
- Cor pulmonale (หัวใจล้มเหลวด้านขวา)
- โรคตับแข็ง (แผลเป็นจากการทำงานของตับ)
เนื่องจาก CF ทำให้เกิดการบาดเจ็บต่อเซลล์และเนื้อเยื่ออย่างต่อเนื่องความเสียหายใด ๆ ที่เกิดกับปอดและอวัยวะอื่น ๆ ส่วนใหญ่จะไม่สามารถย้อนกลับได้ การเสียชีวิตส่วนใหญ่มักเป็นผลจากการหายใจล้มเหลวตามมาด้วยหัวใจล้มเหลวและตับวาย
อาการของ Cystic Fibrosis
สาเหตุ
Cystic fibrosis เกิดจากการกลายพันธุ์ของยีน cystic fibrosis transmembrane receptor (CFTR) ซึ่งมีหน้าที่ในการผลิตโปรตีน CFTR ซึ่งเป็นโปรตีนที่ร่างกายต้องการเพื่อควบคุมการไหลของเกลือและน้ำเข้าและออกจากเซลล์ . หากโปรตีนผิดรูปหรือมีข้อบกพร่องอาจทำให้เกิดการขาดน้ำที่ผิวของเซลล์ซึ่งนำไปสู่การข้นของเมือกโดยรอบ
CF เป็นโรคถอยอัตโนมัติซึ่งหมายความว่าคุณจำเป็นต้องสืบทอดการกลายพันธุ์ของ CFTR จากทั้งแม่และพ่อของคุณเพื่อที่จะเป็นโรคนี้ หากคุณได้รับยีนที่บกพร่องเพียงยีนเดียวคุณจะไม่มี CF แต่จะเป็นพาหะของยีนที่กลายพันธุ์แทน
คุณสามารถถ่ายทอดโรคได้หากพ่อแม่ของคุณแต่ละคนมีการกลายพันธุ์ของ CFTR หรือ CF เอง หากทั้งพ่อและแม่เป็นผู้ให้บริการคุณจะมี:
- โอกาส 25 เปอร์เซ็นต์ที่จะมี CF
- โอกาส 50 เปอร์เซ็นต์ในการเป็นพาหะ
- โอกาส 25 เปอร์เซ็นต์ที่จะไม่ได้รับผลกระทบ
ในทางกลับกันถ้าพ่อแม่คนใดคนหนึ่งของคุณเป็นพาหะและอีกคนมี CF คุณมีโอกาส 50/50 ที่จะมี CF หรือเป็นพาหะ
Cystic fibrosis เป็นหนึ่งในโรคทางพันธุกรรมที่พบบ่อยซึ่งมีผลต่อทารกประมาณหนึ่งในทุกๆ 2,500 คนที่เกิดในสหรัฐอเมริกา
พบได้บ่อยในหมู่คนผิวขาวและเชื้อสายสเปนและเกิดขึ้นน้อยกว่าในคนเชื้อสายแอฟริกันหรือเอเชีย
ปัจจัยเสี่ยงโรคซิสติกไฟโบรซิสการวินิจฉัย
มีการทดสอบบางอย่างที่ใช้ในการวินิจฉัยโรคซิสติกไฟโบรซิส พวกเขาทำงานโดยการตรวจจับการกลายพันธุ์ของ CFTR โดยตรงหรือวัดการเปลี่ยนแปลงทางชีวภาพที่สอดคล้องกับโรคโดยอ้อม วิธีการวินิจฉัยอาจแตกต่างกันไปในระหว่างตั้งครรภ์เมื่อทารกคลอดหรือทุกเวลาหลังจากนั้น
คู่มือการสนทนาเกี่ยวกับ Cystic Fibrosis Doctor
รับคำแนะนำที่พิมพ์ได้ของเราสำหรับการนัดหมายแพทย์ครั้งต่อไปของคุณเพื่อช่วยให้คุณถามคำถามที่ถูกต้อง

จากการทดสอบมาตรฐานสองแบบที่ใช้กันทั่วไปในการวินิจฉัย CF:
- การทดสอบคลอไรด์เหงื่อหรือที่เรียกกันง่ายๆว่าการทดสอบเหงื่อจะวัดปริมาณคลอไรด์บนผิวหนัง เนื่องจาก CF รบกวนการถ่ายเทเกลือเข้าและออกจากเซลล์จึงมีการสะสมของเกลือในเหงื่อ
- การทดสอบ CFTR ทางพันธุกรรม ใช้เพื่อตรวจหาการกลายพันธุ์ที่พบบ่อยที่สุดของการกลายพันธุ์ของ CFTR ในขณะที่มีการกลายพันธุ์ของ CFTR มากกว่า 2,000 รายการที่ทราบว่าก่อให้เกิดโรคปอดเรื้อรัง 23 ชนิดที่รวมอยู่ในแผงมาตรฐานเป็นตัวแทนของผู้ต้องสงสัยที่น่าจะเป็นไปได้มากที่สุด
ในระหว่างตั้งครรภ์การทดสอบทางพันธุกรรม CFTR สามารถใช้เพื่อทดสอบของเหลวที่ได้จากการเจาะน้ำคร่ำหรือเซลล์ที่สกัดผ่านการสุ่มตัวอย่าง chorionic villus (CVS)
การตรวจคัดกรองทารกแรกเกิด นอกจากนี้ยังใช้เป็นมาตรฐานในการวินิจฉัย CF และปัจจุบันได้รับคำสั่งใน 50 รัฐและ District of Columbia สิ่งนี้จะแตกต่างกันไปขึ้นอยู่กับว่าคุณอาศัยอยู่ที่ใดในสหรัฐอเมริกา หากผลการตรวจคัดกรองทารกแรกเกิดเป็นบวกจะใช้การทดสอบเหงื่อเพื่อยืนยันการวินิจฉัย
วิธีการวินิจฉัย Cystic Fibrosisการรักษา
แม้ว่าจะไม่มีวิธีรักษาโรคซิสติกไฟโบรซิส แต่ความก้าวหน้าในการรักษาได้ช่วยยืดอายุของผู้ที่เป็นโรคนี้
จุดมุ่งหมายของการรักษาด้วย CF มีสี่เท่า: เพื่อป้องกันการติดเชื้อรักษาการทำงานของปอดทำให้การย่อยอาหารเป็นปกติและชะลอการลุกลามของโรค
ในบรรดาเครื่องมือบำบัดที่ใช้ในการจัดการ CF:
- เทคนิคการกวาดล้างทางเดินหายใจ (ACTs) จะดำเนินการเพื่อขับไล่และขับเมือกที่สะสมออกจากปอด เทคนิคต่างๆ ได้แก่ การไอหายใจถี่การกระทบหน้าอกหรือการสั่นของผนังหน้าอก
- อาหารไขมันสูงแคลอรีสูง ใช้เพื่อชดเชยการดูดซึมไขมันโปรตีนและสารอาหารในลำไส้
- อาหารเสริมเอนไซม์ตับอ่อน ใช้เพื่อเสริมสร้างเอนไซม์ย่อยอาหารที่ตับอ่อนไม่สามารถผลิตได้เนื่องจากการสะสมของเมือกมากเกินไป
- ยาปฏิชีวนะ รับประทานทุกวันเพื่อป้องกันการติดเชื้อแบคทีเรียในปอด
- Mucolytics- ยาที่ใช้ในการขับเมือกบาง ๆ ก่อนที่จะใช้ ACTs- อาจใช้
- โมดูเลเตอร์ CFTR เป็นยากลุ่มใหม่ที่สามารถแก้ไขข้อบกพร่องบางอย่างในโปรตีน CFTR และฟื้นฟูการทำงานของกฎข้อบังคับ
- การบำบัดด้วยออกซิเจน อาจใช้ในช่วงเฉียบพลันเมื่อการหายใจของคุณบกพร่องอย่างรุนแรง
- สารอาหารทางหลอดเลือดหรือที่เรียกว่าการให้อาหารทางท่ออาจใช้หากคุณไม่สามารถรักษาน้ำหนักได้ด้วยโภชนาการตามปกติ
- การปลูกถ่ายปอด ถือเป็นช่วงที่ปอดของคุณไม่สามารถรองรับการอยู่รอดได้อีกต่อไปหากไม่มีเครื่องช่วยหายใจ
การเผชิญปัญหา
ในปีพ. ศ. 2481 เมื่อโรคซิสติกไฟโบรซิสถูกจัดให้เป็นโรคเป็นครั้งแรกเด็ก ๆ แทบจะไม่ได้ใช้ชีวิตเกินปีแรกของชีวิต ในช่วงทศวรรษที่ 1980 เราคาดว่าจะมีชีวิตอยู่ได้นานถึง 20 ถึง 25 ปี วันนี้ภาพได้เปลี่ยนไปอย่างสิ้นเชิงกับผู้คนที่มีชีวิตที่ดีในช่วงอายุ 40 และ 50 ปีหากการรักษาเริ่มต้นและปฏิบัติตาม
นี่ไม่ได้หมายความว่า CF มีความร้ายแรงน้อยกว่าที่เคยเป็นมา เป็นเหตุการณ์ที่เปลี่ยนแปลงชีวิตต้องอาศัยความขยันหมั่นเพียรและความสม่ำเสมอเพื่อไม่เพียงรับมือกับโรค แต่ดำเนินชีวิตตามมาตรฐานสูงสุดเท่าที่จะเป็นไปได้
ด้วยเหตุนี้คุณจำเป็นต้องทำให้ CF ในชีวิตของคุณเป็นปกติโดยกำหนดกิจวัตรและการปฏิบัติเพื่อหลีกเลี่ยงการขึ้น ๆ ลง ๆ ที่อาจทำให้เกิดความเครียดและเพิ่มความพิการ ในการพิจารณาคุณจะต้อง:
- จัดการโภชนาการของคุณ. คนที่เป็นโรค CF มักต้องการแคลอรี่ต่อวันเป็นสองเท่าของคนอื่น ๆ
- ออกกำลังกายสม่ำเสมอ กิจวัตรการออกกำลังกายควรเกี่ยวข้องกับกิจกรรมแอโรบิกอย่างน้อย 20 ถึง 30 นาทีสามครั้งต่อสัปดาห์ ค้นหาสิ่งที่น่าสนุกที่คุณสามารถทำได้ตลอดชีวิต
- ให้ความชุ่มชื้น. การทำเช่นนี้ช่วยให้ปอดและลำไส้ทำงานได้อย่างถูกต้อง คุณควรดื่มน้ำปริมาณสูงไม่น้อยกว่าหกถึงแปดแก้วต่อวันทั้งนี้ขึ้นอยู่กับอายุของคุณ
- ดำเนินการทางเดินหายใจอย่างถูกต้อง เนื่องจากความต้องการด้านสุขภาพของคุณเปลี่ยนไปดังนั้นประเภทของเครื่องมือในการกวาดล้างที่คุณต้องการก็เช่นกัน พูดคุยกับแพทย์โรคปอดหรือนักกายภาพบำบัดของคุณหากคุณไม่บรรลุผลที่ควรจะเป็น
- ขอความช่วยเหลือ นอกจากเพื่อนและครอบครัวแล้วคุณสามารถติดต่อบทที่ใกล้ที่สุดของมูลนิธิ Cystic Fibrosis (CFF) เพื่อเชื่อมต่อกับเครือข่ายสนับสนุนในพื้นที่ของคุณ
- ขอความช่วยเหลือทางการเงิน CFF นำเสนอบริการที่ช่วยให้ครอบครัวรับมือกับค่ารักษา CF ที่มีราคาสูงได้ดีขึ้น
คำจาก Verywell
ในขณะที่การตรวจคัดกรองทารกแรกเกิดทำให้อัตราการวินิจฉัย CF ในทารกเพิ่มขึ้นอย่างมาก แต่การวินิจฉัยมากกว่า 25 เปอร์เซ็นต์จะเกิดขึ้นในช่วงวัยเด็กวัยรุ่นและผู้ใหญ่ตอนต้นเท่านั้น
นี่เป็นปัญหาเนื่องจากการวินิจฉัยและการรักษาในระยะเริ่มต้นสามารถป้องกันภาวะแทรกซ้อนที่รุนแรงมากขึ้นของ CF ก่อนที่จะเกิดความเสียหายร้ายแรง แม้ว่าการรักษาจะไม่สามารถหยุดหรือย้อนกลับของโรคได้ แต่ก็สามารถมั่นใจได้ว่าจะปลอดโรคได้อีกหลายปี
ด้วยเหตุนี้สิ่งสำคัญคือต้องทราบอาการเริ่มต้นของ CF และปรึกษาแพทย์หากคุณสงสัยว่าบุตรหลานของคุณอาจเป็นโรค นี่เป็นความจริงโดยเฉพาะอย่างยิ่งในรัฐที่ตรวจคัดกรองด้วยการตรวจเลือด IRT เท่านั้นซึ่งอาจส่งผลให้เด็กจำนวนมากถึง 5 เปอร์เซ็นต์ได้รับการวินิจฉัยที่ล่าช้าหรือผลลัพธ์ที่ผิดพลาดตามการวิจัยจากคณะแพทยศาสตร์และสาธารณสุขแห่งมหาวิทยาลัยวิสคอนซิน .
อาการอะไรที่คุณสามารถคาดหวังได้จาก Cystic Fibrosis?